Congenital Diaphragmatic Hernia
Neonatal Disease
Complete post test after you read this module. Save your certificate of completion!
Congenital Diaphragmatic Hernia
Overview:
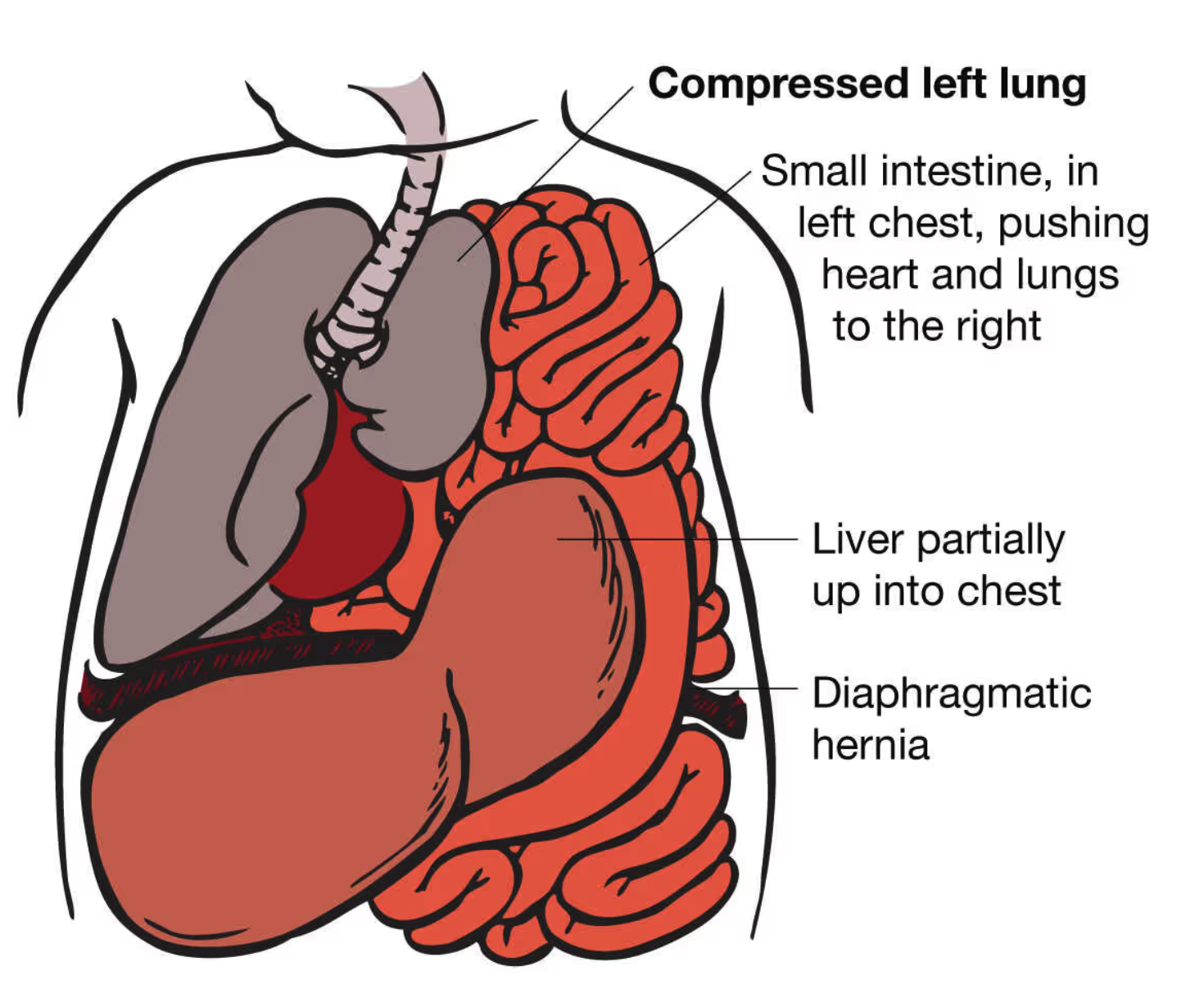
Congenital Diaphragmatic Hernia (CDH) is a developmental defect characterized by the presence of an opening in the diaphragm, the muscle that separates the chest cavity from the abdominal cavity. This opening allows abdominal organs, such as the stomach, liver, and intestines, to move into the chest cavity, hindering normal lung development.
1. Pathophysiology:
Congenital diaphragmatic hernia (CDH) occurs when there is a defect in the diaphragm, the muscle that separates the chest cavity from the abdominal cavity, allowing abdominal organs like the stomach, intestines, and sometimes the liver to herniate into the chest. This defect disrupts normal lung development, leading to pulmonary hypoplasia (underdeveloped lungs), which is a hallmark of CDH. As the herniated organs occupy space in the chest, they compress the developing lungs, reducing lung volume and impairing lung expansion at birth. Additionally, the condition is often associated with pulmonary hypertension due to vascular changes in the lungs, where blood vessels constrict in response to low oxygen levels. This further exacerbates respiratory difficulties, increasing the risk of hypoxemia and respiratory failure. The severity of CDH depends on the size of the diaphragmatic defect, the degree of lung hypoplasia, and the presence of pulmonary hypertension, which together contribute to the neonatal morbidity and mortality associated with the condition.
2. Clinical Presentation:
The severity of CDH can vary, and symptoms depend on the extent of lung
compression. Common clinical features include:
Respiratory distress shortly after birth.
Cyanosis (bluish discoloration of the skin and mucous membranes).
Tachypnea (rapid breathing).
Barrel-shaped chest due to increased work of breathing.
Bowel sounds heard in the chest.
3. Diagnostic Evaluation:
Prenatal Ultrasound: CDH is often diagnosed during routine prenatal ultrasounds, allowing for early preparation and planning.
Chest X-ray: After birth, a chest X-ray is performed to confirm the diagnosis and assess the extent of lung involvement.
Echocardiogram: As CDH is associated with other congenital anomalies, an echocardiogram is often conducted to evaluate heart function.
4. Prognosis:
The prognosis for CDH depends on various factors, including the size of the diaphragmatic defect, the degree of pulmonary hypoplasia, and the presence of associated anomalies. Despite advances in medical care, CDH can be a life-threatening condition, especially in cases of severe pulmonary hypoplasia.
5. Long-Term Considerations:
Survivors of CDH may face long-term respiratory issues, and ongoing medical follow-up is essential. Some children may require respiratory support or surgery for complications associated with CDH.
6. Chest X-ray Findings in Congenital Diaphragmatic Hernia (CDH):
Chest X-rays play a crucial role in the diagnosis and evaluation of congenital diaphragmatic hernia (CDH). The findings on a chest X-ray can provide important information about the extent of lung involvement and the positioning of abdominal organs within the chest cavity. Here are the typical chest X-ray findings associated with CDH:
Visceral Herniation: The primary characteristic seen on a chest X-ray in CDH is the presence
of abdominal organs, such as the stomach, liver, and intestines, within the chest cavity.
These organs typically appear as a soft-tissue mass occupying the thoracic space.
Shift of Mediastinum: The presence of abdominal contents in the chest can cause a shift
in the mediastinum (the central partition of the thoracic cavity). This shift may lead to
compression of the lung on the affected side, contributing to pulmonary hypoplasia.
Opacity in Thoracic Cavity: The herniated abdominal organs may appear as an opaque
or soft-tissue density within the thoracic cavity on the X-ray. This opacity contrasts with
the air-filled lungs, aiding in the identification of the herniated contents.
Contralateral Lung Hyperinflation: Due to the compression of the affected lung, the
contralateral lung (lung on the opposite side) may appear hyperinflated. This hyperinflation
is a compensatory response to the reduced lung capacity on the side with CDH.
Cardiac Deviation: The position of the heart may be altered due to the displacement caused
by the herniated abdominal organs. The heart may appear shifted toward the opposite side.
Barrel-Shaped Chest: In severe cases of CDH, the chest may exhibit a barrel-shaped
appearance on the X-ray. This is a result of increased work of breathing and respiratory distress.
7. Intervention
Because of associated persistent pulmonary hypertension of the newborn (PPHN) and pulmonary hypoplasia, medical therapy in patients with congenital diaphragmatic hernia (CDH) is directed toward optimizing oxygenation while avoiding barotrauma.
Delivery Room Management:
Place a vented orogastric tube and connect it to continuous suction to prevent bowel distension and further lung compression.
Avoid mask ventilation; immediately intubate the trachea to prevent lung compression.
Be cautious with high peak inspiratory pressures, and watch for early pneumothorax if stabilization is not achieved.
Surfactant Use:
CDH infants may have surfactant deficiency, but administering exogenous surfactant does not improve survival or long-term outcomes.
Ventilation Strategies:
Mechanical ventilation aims to avoid high inspiratory pressures and synchronize with the infant's respiratory effort.
High-frequency oscillatory ventilation (HFOV) may be considered at experienced centers to avoid high inspiratory pressures.
Metabolic Support:
Maintain glucose and ionized calcium concentrations within the reference range.
Support blood pressure using volume expansion and inotropic agents.
Oxygen and Carbon Dioxide Targets:
Controversy exists regarding the appropriate PaO2 and PaCO2 targets.
PaO2 concentrations >50 mm Hg are generally considered adequate; aiming for higher levels may lead to increased ventilator support.
The approach to PaCO2 maintenance (low for pulmonary vasodilation, permissive hypercapnia, or normocarbia) is debated without clear consensus.
Inhaled Nitric Oxide (iNO):
iNO has revolutionized persistent pulmonary hypertension of the newborn (PPHN) treatment but is controversial in CDH.
iNO does not reduce mortality or ECMO need in CDH but may stabilize infants with critical hypoxemia.
Caution is advised if ECMO is not immediately available, and recent studies explore long-term low-dose iNO therapy for late or recurrent pulmonary hypertension.
9. Surgical Repair:
Surgical repair of a congenital diaphragmatic hernia (CDH) is a critical intervention aimed at closing the defect in the diaphragm and preventing further complications related to the hernia. Here's an overview of the procedure and key considerations:
Timing of Surgery:
Surgery is typically performed within the first 48 to 72 hours of life once the neonate has been stabilized, particularly if they are on mechanical ventilation or high-frequency ventilation.
Pre-surgical stabilization may involve managing pulmonary hypertension, oxygenation, and fluid balance. Some infants may require ECMO (extracorporeal membrane oxygenation) if pulmonary function is severely impaired.
Surgical Procedure:
Incision: A midline abdominal or thoraco-abdominal incision is made to access the chest and repair the defect.
Reduction of the hernia: The herniated abdominal contents (e.g., stomach, intestines, liver) are gently reduced back into the abdomen.
Closing the defect: The defect in the diaphragm is then sutured closed. In some cases, if the defect is too large, a synthetic patch or muscle flap may be used to cover the defect and reinforce the diaphragm.
Challenges in Surgery:
Lung hypoplasia: Many neonates with CDH have underdeveloped lungs, and the surgeon must ensure that the lungs are adequately expanded and functioning after the hernia is reduced.
Pulmonary hypertension: Preoperative and postoperative management of pulmonary hypertension is crucial. It can persist after surgery and requires treatment with medications like inhaled nitric oxide or sildenafil to reduce pressure in the pulmonary arteries.
Gastric decompression: During the surgery, gastric decompression via a nasogastric tube is essential to reduce the risk of gastric distention, which can worsen respiratory function.
Post-Surgical Care:
Intensive monitoring in the NICU is necessary, especially for signs of respiratory distress, infection, or failure to thrive.
Ventilatory support may be required for some time after surgery, as the neonate's lungs recover and adjust to the new respiratory demands.
Feeding and growth may need to be carefully managed post-surgery, as some infants with CDH may have delayed or impaired feeding.
Long-Term Outlook:
Lung function may remain compromised in some neonates due to the effects of pulmonary hypoplasia or chronic lung disease (e.g., bronchopulmonary dysplasia).
Follow-up care is essential to monitor for developmental delays, lung function, and nutritional support, as many infants with CDH may require ongoing respiratory therapy or interventions as they grow.
Why is it important for an RT to know about Congenital Diaphragmatic Hernia ?
Congenital diaphragmatic hernia (CDH) is one of the highest-stakes deliveries a NICU respiratory therapist will attend because the first few minutes can prevent rapid decompensation and set the tone for survival. These babies are born with lung hypoplasia and significant pulmonary hypertension risk, so you should expect severe respiratory distress, low oxygenation despite support, and rapid worsening if the stomach/abdominal contents inflate in the chest. At delivery, the priority is immediate airway control and gentle ventilation: avoid bag-mask ventilation when possible, intubate early, place an orogastric (OG) tube to continuous suction to decompress the stomach, and use lung-protective settings (low pressures, permissive hypercapnia as ordered) to minimize barotrauma to fragile hypoplastic lungs. Be ready for high oxygen needs, careful titration to ordered targets (often pre/post-ductal monitoring), and escalation to advanced modes (HFV/HFOV) and inhaled nitric oxide if pulmonary hypertension is suspected or confirmed—while recognizing that some infants will remain refractory and need ECMO evaluation/activation depending on your center’s criteria. RTs also need to anticipate a coordinated team workflow (lines, sedation, labs, echo), frequent blood gases, and ongoing vigilance for air leaks and hemodynamic instability—because in CDH, “more pressure” can be harmful, and smart, controlled respiratory management is a major determinant of outcomes.